 中文网站
中文网站
Aux premières heures du 29 décembre, le NEJM a publié en ligne une nouvelle étude clinique de phase III sur le nouveau coronavirus chinois VV116. Les résultats ont montré que le VV116 n'était pas pire que le Paxlovid (nématovir/ritonavir) en termes de durée de guérison clinique et qu'il présentait moins d'effets indésirables.

Source de l'image : NEJM
Durée médiane de convalescence : 4 jours, taux d’événements indésirables : 67,4 %
Le VV116 est un médicament nucléosidique oral contre le nouveau coronavirus (SARS-CoV-2) développé en collaboration avec Junsit et Wang Shan Wang Shui, et est un inhibiteur de la RdRp avec le remdesivir de Gilead, le molnupiravir de Merck Sharp & Dohme et l'azelvudine de Real Biologics.
En 2021, un essai clinique de phase II du VV116 a été mené à terme en Ouzbékistan. Les résultats de l'étude ont montré que le groupe traité par VV116 présentait une amélioration des symptômes cliniques et une réduction significative du risque d'évolution vers une forme grave et de décès, comparativement au groupe témoin. Forts de ces résultats positifs, le VV116 a été approuvé en Ouzbékistan pour le traitement des patients atteints de COVID-19 modérée à sévère et est devenu le premier nouveau médicament oral contre les maladies coronariennes autorisé à la vente à l'étranger, en Chine [1].
Cet essai clinique de phase III[2] (NCT05341609), mené par le Pr Zhao Ren de l'hôpital Ruijin de Shanghai, le Pr Gaoyuan de l'hôpital Renji de Shanghai et l'académicien Ning Guang de l'hôpital Ruijin de Shanghai, a été réalisé pendant l'épidémie causée par le variant Omicron (B.1.1.529) de mars à mai à Shanghai. Son objectif était d'évaluer l'efficacité et l'innocuité du VV116 par rapport au Paxlovid pour le traitement précoce des patients atteints de COVID-19 légère à modérée.

Source de l'image : Référence 2
Un essai multicentrique, randomisé, contrôlé et mené en simple aveugle auprès de 822 patients adultes atteints de Covid-19 présentant un risque élevé d'aggravation et des symptômes légers à modérés a été réalisé entre le 4 avril et le 2 mai 2022 afin d'évaluer l'éligibilité des participants dans sept hôpitaux de Shanghai, en Chine. Au final, 771 participants ont reçu soit du VV116 (384 patients, 600 mg toutes les 12 heures le premier jour et 300 mg toutes les 12 heures les jours 2 à 5), soit du Paxovid (387 patients, 300 mg de nimatuvir + 100 mg de ritonavir toutes les 12 heures pendant 5 jours) par voie orale.
Les résultats de cette étude clinique ont montré que le traitement précoce par VV116 pour les cas de COVID-19 légers à modérés a atteint le critère d'évaluation principal (délai de rétablissement clinique durable) prévu par le protocole clinique : le délai médian de rétablissement clinique était de 4 jours dans le groupe VV116 et de 5 jours dans le groupe Paxlovid (rapport de risque, 1,17 ; IC à 95 %, 1,02 à 1,36 ; limite inférieure > 0,8).
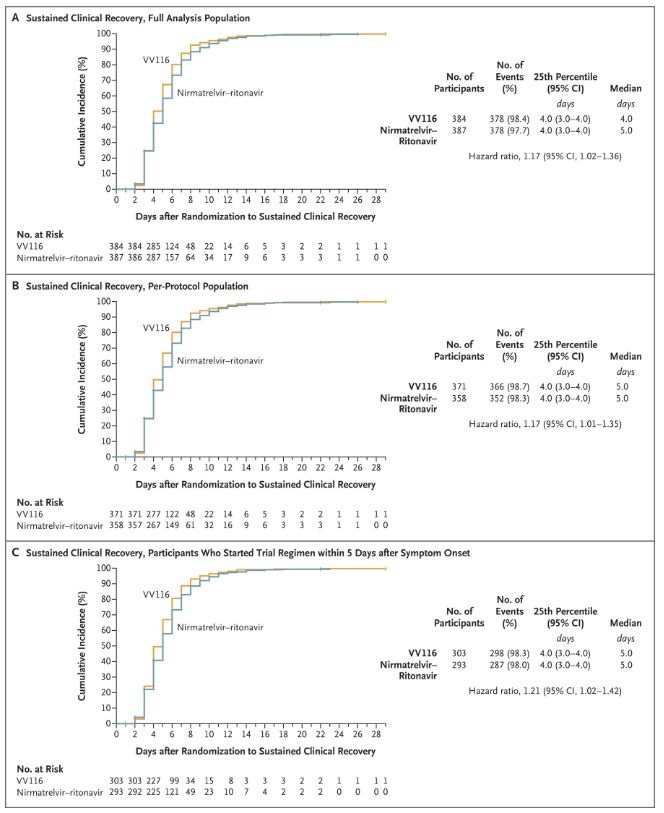
Maintenir le temps de récupération clinique
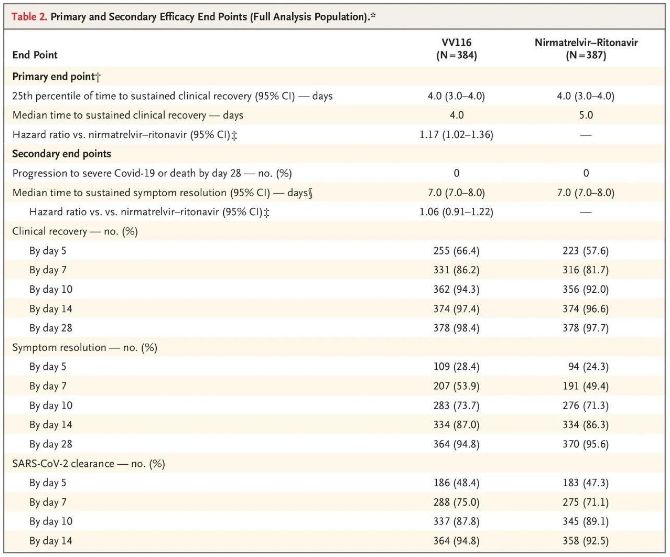
Critères d’évaluation principaux et secondaires de l’efficacité (analyse exhaustive de la population)
Source de l'image : Référence 2
En termes de sécurité, les participants recevant le VV116 ont signalé moins d'événements indésirables (67,4 %) que ceux recevant le Paxlovid (77,3 %) lors du suivi de 28 jours, et l'incidence des événements indésirables de grade 3/4 était plus faible pour le VV116 (2,6 %) que pour le Paxlovid (5,7 %).
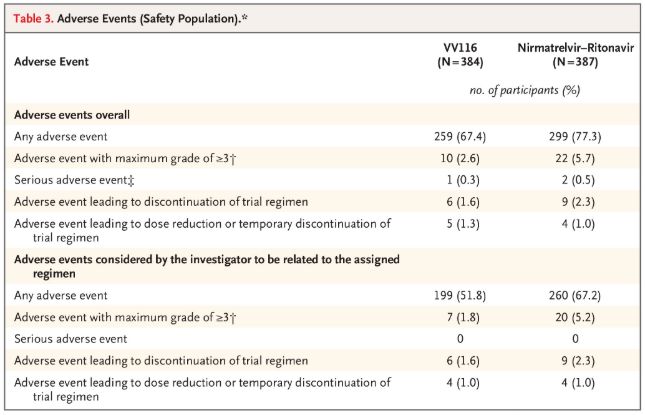
Événements indésirables (personnes en sécurité)
Source de l'image : Référence 2
Controverses et questions
Le 23 mai 2022, Juniper a annoncé que l'étude clinique d'enregistrement de phase III du VV116 par rapport au PAXLOVID pour le traitement précoce du COVID-19 léger à modéré (NCT05341609) avait atteint son objectif principal.

Source de l'image : Référence 1
À une époque où les détails de l'essai faisaient défaut, la controverse entourant l'étude de phase III était double : premièrement, il s'agissait d'une étude en simple aveugle et, en l'absence de groupe témoin placebo, on craignait qu'il soit difficile de juger le médicament de manière totalement objective ; deuxièmement, des questions se posaient quant aux critères d'évaluation cliniques.
Les critères d'inclusion clinique de l'étude Juniper sont : (i) un résultat positif au test de dépistage du nouveau coronavirus, (ii) un ou plusieurs symptômes légers ou modérés de la COVID-19, et (iii) un risque élevé de forme grave de la COVID-19, pouvant entraîner le décès. Toutefois, le seul critère d'évaluation principal est le délai d'obtention d'une guérison clinique durable.
Juste avant l’annonce, le 14 mai, Juniper avait révisé les critères d’évaluation cliniques en supprimant l’un des critères d’évaluation cliniques principaux, « la proportion de conversions en maladie grave ou en décès » [3].
![]()
Source de l'image : Référence 1
Ces deux principaux points de désaccord ont également été spécifiquement abordés dans l'étude publiée.
En raison de l'apparition soudaine d'Omicron, la production des comprimés placebo pour Paxlovid n'a pas pu être achevée avant le début de l'essai. Par conséquent, les investigateurs n'ont pas pu mener cet essai en double aveugle avec double placebo. Concernant le volet simple aveugle de l'essai clinique, Juniper a indiqué que le protocole avait été élaboré après concertation avec les autorités réglementaires et que, de ce fait, ni l'investigateur (y compris l'évaluateur du critère d'évaluation principal) ni le promoteur ne connaîtraient l'attribution spécifique du traitement jusqu'au verrouillage de la base de données finale à la fin de l'étude.
Jusqu'à l'analyse finale, aucun décès ni aggravation de la Covid-19 n'a été constaté chez les participants à l'essai. Par conséquent, aucune conclusion ne peut être tirée quant à l'efficacité du vaccin VV116 pour prévenir l'évolution vers une forme grave ou critique de la Covid-19, ni le décès. Les données indiquent que le délai médian estimé entre la randomisation et la régression durable des symptômes cibles liés à la Covid-19 était de 7 jours (IC à 95 %, 7 à 8) dans les deux groupes (risque relatif : 1,06 ; IC à 95 %, 0,91 à 1,22) [2]. Il est aisé de comprendre pourquoi le critère d'évaluation principal, « taux de conversion vers une forme grave ou de décès », initialement défini avant la fin de l'essai, a été abandonné.
Le 18 mai 2022, la revue Emerging Microbes & Infections a publié les résultats du premier essai clinique du VV116 chez des patients infectés par la variante Omicron [4], une étude de cohorte ouverte et prospective avec 136 patients hospitalisés confirmés.
Les données de l'étude ont montré que chez les patients infectés par Omicron ayant reçu le vaccin VV116 dans les 5 jours suivant leur premier test d'amplification des acides nucléiques positif, le délai de régression de l'amplification était de 8,56 jours, inférieur aux 11,13 jours observés dans le groupe témoin. L'administration de VV116 aux patients symptomatiques dans le délai imparti par l'étude (2 à 10 jours après le premier test d'amplification des acides nucléiques positif) a réduit ce délai chez tous les patients. Concernant la sécurité du médicament, aucun effet indésirable grave n'a été observé dans le groupe traité par VV116.
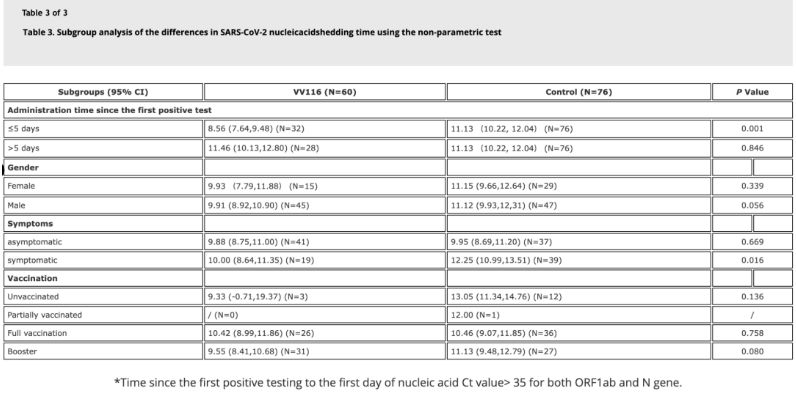
Source de l'image : Référence 4
Trois essais cliniques sont actuellement en cours sur le VV116, dont deux sont des études de phase III portant sur les cas de COVID-19 légers à modérés (NCT05242042 et NCT05582629). Le troisième essai, mené auprès de patients atteints de COVID-19 modérés à sévères, est une étude clinique internationale multicentrique, randomisée, en double aveugle de phase III (NCT05279235) visant à évaluer l'efficacité et l'innocuité du VV116 par rapport au traitement standard. Selon l'annonce de Juniper, le premier patient a été inclus dans l'étude et a reçu le traitement en mars 2022.

Source de l'image : clinicaltrials.gov
Références :
[1]Junshi Biotech : Annonce concernant le critère d'évaluation principal de l'étude clinique de phase III enregistrée comparant VV116 à PAXLOVID pour le traitement précoce des cas légers à modérés de COVID-19
[2]https://www.nejm.org/doi/full/10.1056/NEJMoa2208822?query=featured_home[3]https://clinicaltrials.gov/ct2/show/record/NCT05341609[4] Ensi Ma, Jingwen Ai, Yi Zhang, Jianming Zheng, Xiaogang Gao, Junming Xu, Hao Yin, Zhiren Fu, Hao Xing, Li Li, Liying Sun, Heyu Huang, Quanbao Zhang, Linlin Xu, Yanting Jin, Rui Chen, Guoyue Lv, Zhijun Zhu, Wenhong Zhang, Zhengxin Wang. (2022) Profil des infections à Omicron et statut vaccinal chez 1881 receveurs de transplantation hépatique : une cohorte rétrospective multicentrique. Microbes et infections émergents 11 : 1, pages 2636-2644.
Date de publication : 6 janvier 2023
 Paramètres de confidentialité
Paramètres de confidentialité