 中文网站
中文网站
Nat Med | Une approche multi-omique pour cartographier le paysage tumoral, immunitaire et microbien intégré du cancer colorectal révèle l'interaction du microbiome avec le système immunitaire
Bien que les biomarqueurs du cancer colorectal primitif aient fait l'objet de nombreuses études ces dernières années, les recommandations cliniques actuelles s'appuient uniquement sur la classification TNM (Tumeur-Ganglions-Métastases) et la détection d'anomalies du système de réparation des mésappariements de l'ADN (MMR) ou d'une instabilité microsatellitaire (MSI) (en complément des analyses histopathologiques standard) pour déterminer les recommandations thérapeutiques. Les chercheurs ont constaté une absence de corrélation entre les réponses immunitaires, évaluées par l'expression génique, les profils microbiens et le stroma tumoral dans la cohorte de patients atteints de cancer colorectal du TCGA (The Cancer Genome Atlas) et la survie des patients.
Au fil des progrès de la recherche, il a été démontré que les caractéristiques quantitatives du cancer colorectal primitif, notamment la nature cellulaire, immunitaire, stromale ou microbienne du cancer, sont significativement corrélées aux résultats cliniques, mais la compréhension de la manière dont leurs interactions affectent l'évolution des patients reste limitée.
Pour analyser la relation entre la complexité phénotypique et le pronostic, une équipe de chercheurs de l'Institut Sidra de recherche médicale au Qatar a récemment développé et validé un score intégré (mICRoScore) permettant d'identifier un groupe de patients présentant de bons taux de survie. Ce score combine les caractéristiques du microbiome et les constantes de rejet immunitaire (ICR). L'équipe a réalisé une analyse génomique complète d'échantillons frais congelés provenant de 348 patients atteints d'un cancer colorectal primitif. Cette analyse comprenait le séquençage de l'ARN des tumeurs et des tissus colorectaux sains appariés, le séquençage de l'exome entier, le séquençage profond des gènes du récepteur des lymphocytes T et de l'ARNr bactérien 16S, complété par le séquençage du génome tumoral entier afin de mieux caractériser le microbiome. L'étude a été publiée dans Nature Medicine sous le titre « Atlas intégré des tumeurs, du système immunitaire et du microbiome du cancer du côlon ».

Article publié dans Nature Medicine
Présentation d'AC-ICAM
Des chercheurs ont utilisé une plateforme génomique orthogonale pour analyser des échantillons tumoraux congelés et des tissus coliques sains adjacents (paires tumeur-tissu sain) provenant de patients atteints d'un cancer du côlon diagnostiqué histologiquement et n'ayant pas reçu de traitement systémique. Après séquençage de l'exome entier (WES), contrôle de la qualité des données de séquençage d'ARN (RNA-seq) et vérification des critères d'inclusion, les données génomiques de 348 patients ont été conservées et utilisées pour des analyses ultérieures, avec un suivi médian de 4,6 ans. L'équipe de recherche a nommé cette ressource Sidra-LUMC AC-ICAM : une carte et un guide des interactions entre le système immunitaire, le cancer et le microbiome (Figure 1).
Classification moléculaire utilisant l'ICR
En identifiant un ensemble modulaire de marqueurs génétiques immunitaires pour une immunosurveillance continue du cancer, appelé constante immunitaire de rejet (ICR), l'équipe de recherche a optimisé l'ICR en la réduisant à un panel de 20 gènes couvrant différents types de cancers, dont le mélanome, le cancer de la vessie et le cancer du sein. L'ICR a également été associée à la réponse à l'immunothérapie dans divers types de cancers, notamment le cancer du sein.
Dans un premier temps, les chercheurs ont validé la signature ICR de la cohorte AC-ICAM, en utilisant une approche de co-classification basée sur les gènes ICR afin de classer la cohorte en trois clusters/sous-types immunitaires : ICR élevé (tumeurs « chaudes »), ICR moyen et ICR faible (tumeurs « froides ») (Figure 1b). Ils ont ensuite caractérisé la propension immunitaire associée aux sous-types moléculaires consensuels (CMS), une classification du cancer du côlon basée sur le transcriptome. Les catégories CMS comprenaient CMS1/immunitaire, CMS2/canonique, CMS3/métabolique et CMS4/mésenchymateux. L’analyse a montré que les scores ICR étaient corrélés négativement avec certaines voies de signalisation des cellules cancéreuses dans tous les sous-types CMS, et que des corrélations positives avec les voies immunosuppressives et stromales n’étaient observées que dans les tumeurs CMS4.
Dans tous les CMS, l'abondance des sous-ensembles de cellules tueuses naturelles (NK) et de cellules T était la plus élevée dans les sous-types immunitaires à ICR élevé, avec une plus grande variabilité dans les autres sous-ensembles de leucocytes (Figure 1c). Les sous-types immunitaires ICR présentaient une OS et une PFS différentes, avec une augmentation progressive de l'ICR de faible à élevé (Figure 1d), validant le rôle pronostique de l'ICR dans le cancer colorectal.
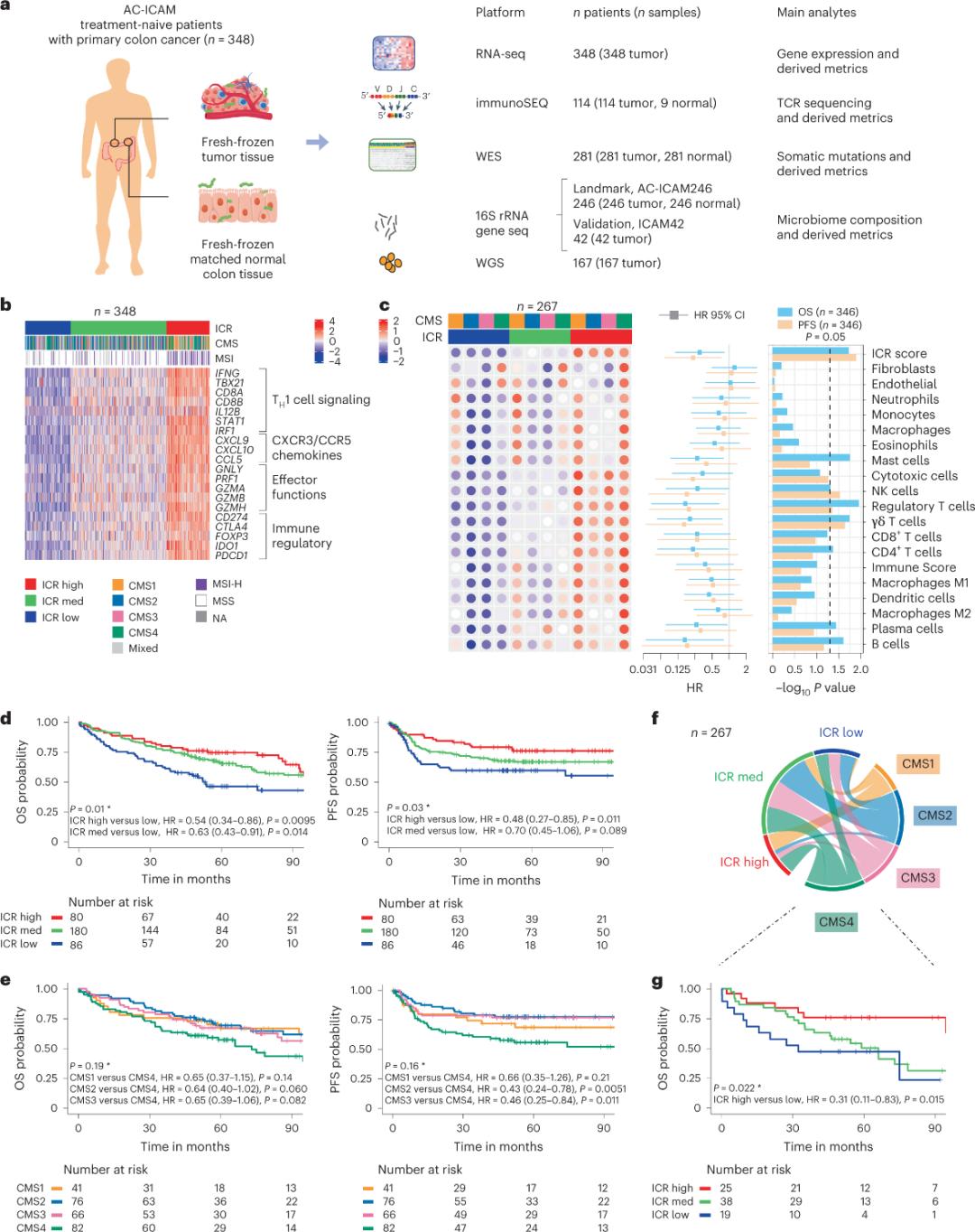
Figure 1. Conception de l'étude AC-ICAM, signature génétique liée à l'immunité, sous-types immunitaires et moléculaires et survie.
ICR capture les lymphocytes T enrichis en cellules tumorales et amplifiés clonalement
Seule une minorité de lymphocytes T infiltrant le tissu tumoral sont spécifiques des antigènes tumoraux (moins de 10 %). Par conséquent, la majorité des lymphocytes T intratumoraux sont qualifiés de lymphocytes T non spécifiques. La plus forte corrélation avec le nombre de lymphocytes T conventionnels possédant des TCR fonctionnels a été observée dans les sous-populations de cellules stromales et de leucocytes (détectées par RNA-seq), permettant d'estimer les sous-populations de lymphocytes T (Figure 2a). Dans les clusters ICR (classification globale et CMS), la clonalité la plus élevée des TCR SEQ immunitaires a été observée dans les groupes ICR-élevé et de sous-type CMS1/immunitaire (Figure 2c), avec la plus forte proportion de tumeurs ICR-élevé. L’analyse du transcriptome complet (18 270 gènes) a révélé que six gènes ICR (IFNG, STAT1, IRF1, CCL5, GZMA et CXCL10) figuraient parmi les dix gènes les plus fortement associés à la clonalité du TCR (ImmunoSEQ) (Figure 2d). La clonalité du TCR (ImmunoSEQ) était plus fortement corrélée à la plupart des gènes ICR que les corrélations observées avec les marqueurs CD8+ spécifiques des tumeurs (Figures 2f et 2g). En conclusion, cette analyse suggère que la signature ICR reflète la présence de lymphocytes T enrichis dans la tumeur et ayant subi une amplification clonale, ce qui pourrait expliquer sa valeur pronostique.
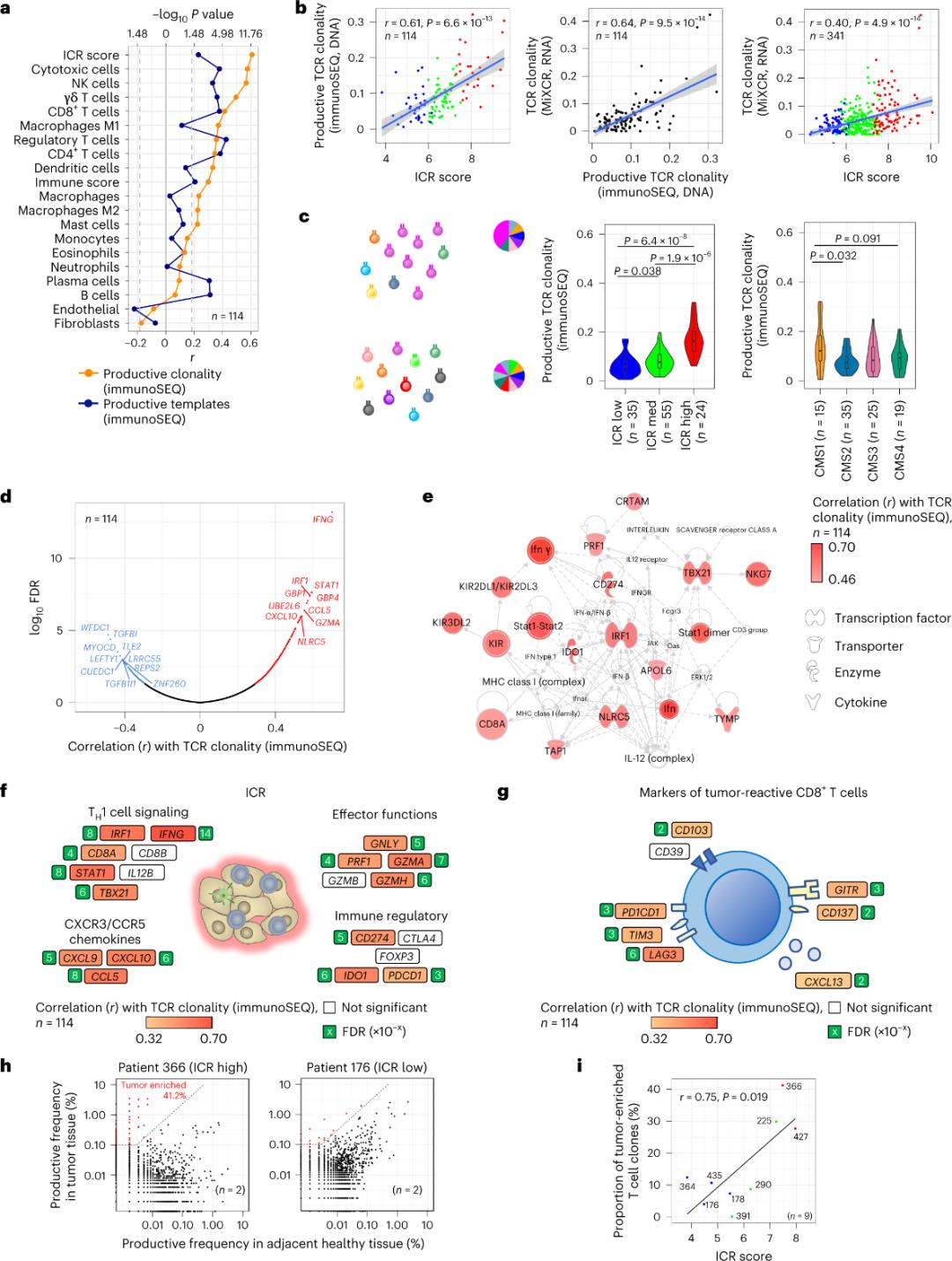
Figure 2. Métriques TCR et corrélation avec les gènes liés à l'immunité, les sous-types immunitaires et moléculaires.
Composition du microbiome dans les tissus sains et cancéreux du côlon
Les chercheurs ont réalisé le séquençage de l'ARNr 16S à partir d'ADN extrait de tissus tumoraux et de tissus coliques sains appariés provenant de 246 patients (Figure 3a). À des fins de validation, ils ont également analysé les données de séquençage du gène de l'ARNr 16S de 42 échantillons tumoraux supplémentaires pour lesquels aucun ADN normal apparié n'était disponible. Ils ont d'abord comparé l'abondance relative de la flore entre les tumeurs et les tissus coliques sains appariés. Clostridium perfringens était significativement plus abondant dans les tumeurs que dans les échantillons sains (Figures 3a à 3d). Aucune différence significative n'a été observée concernant la diversité alpha (diversité et abondance des espèces dans un échantillon donné) entre les tumeurs et les tissus sains, et une légère réduction de la diversité microbienne a été constatée dans les tumeurs à ICR élevé par rapport aux tumeurs à ICR faible.
Afin de détecter des associations cliniquement pertinentes entre les profils microbiens et les résultats cliniques, les chercheurs ont utilisé les données de séquençage du gène de l'ARNr 16S pour identifier les caractéristiques du microbiome prédictives de la survie. Lors de l'étude AC-ICAM246, ils ont appliqué un modèle de régression de Cox OS qui a sélectionné 41 caractéristiques présentant des coefficients non nuls (associées à un risque de mortalité différentiel), appelées classificateurs MBR (Figure 3f).
Dans cette cohorte d'entraînement (ICAM246), un score MBR faible (MBR < 0, MBR faible) était associé à un risque de décès significativement plus faible (85 %). Les chercheurs ont confirmé l'association entre un faible score MBR (risque) et une survie globale prolongée dans deux cohortes validées indépendamment (ICAM42 et TCGA-COAD) (Figure 3). L'étude a montré une forte corrélation entre les cocci endogastriques et les scores MBR, similaires dans les tissus tumoraux et les tissus sains du côlon.
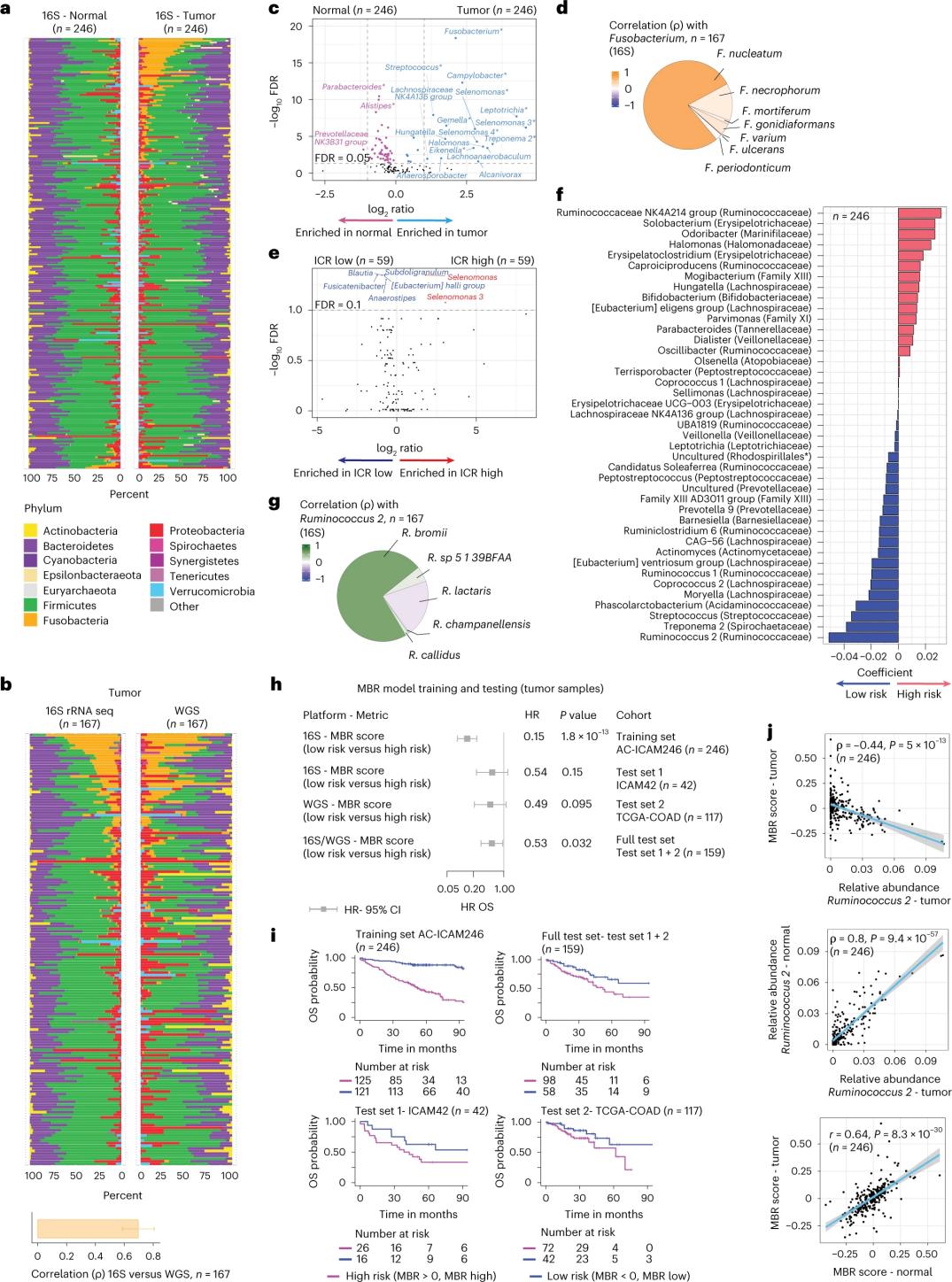
Figure 3. Microbiome dans les tissus tumoraux et sains et relation avec l'ICR et la survie des patients.
Conclusion
L'approche multi-omique utilisée dans cette étude permet une détection et une analyse approfondies de la signature moléculaire de la réponse immunitaire dans le cancer colorectal et révèle l'interaction entre le microbiome et le système immunitaire. Le séquençage profond du TCR des tissus tumoraux et sains a révélé que l'effet pronostique de l'ICR pourrait être dû à sa capacité à capturer des clones de lymphocytes T enrichis dans la tumeur et possiblement spécifiques d'antigènes tumoraux.
En analysant la composition du microbiome tumoral par séquençage du gène ARNr 16S dans des échantillons AC-ICAM, l'équipe a identifié une signature microbienne (score de risque MBR) à forte valeur pronostique. Bien que cette signature provienne d'échantillons tumoraux, une forte corrélation a été observée entre le score de risque MBR du côlon sain et celui de la tumeur, suggérant qu'elle pourrait refléter la composition du microbiome intestinal des patients. La combinaison des scores ICR et MBR a permis d'identifier et de valider un biomarqueur étudiant multi-omique prédictif de la survie chez les patients atteints d'un cancer du côlon. L'ensemble de données multi-omiques de cette étude constitue une ressource précieuse pour une meilleure compréhension de la biologie du cancer du côlon et contribue à la découverte d'approches thérapeutiques personnalisées.
Date de publication : 15 juin 2023
 Paramètres de confidentialité
Paramètres de confidentialité